FDA承认中断的通知召回公司在大流行期间,虽然监管专家指出检查和程序越来越少。
FDA承认中断的通知召回公司在大流行期间,虽然监管专家指出检查和程序越来越少。
食品药品监督管理局医疗器械产品召回去年降至2013年以来的最低水平,显示医疗设计与外包记得来自监管机构的数据分析。
医学技术管理专家说COVID-19流行显示,FDA监管改进的空间和时间监控的医疗设备。
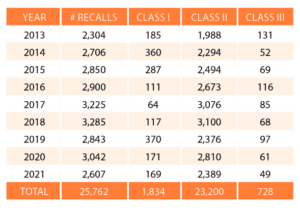
设备产品召回2021财年(截至2021年9月30日)总计2607人,减少了14.3%的3042年回忆2020年财政报告。
设备回忆发生的下降同时FDA更加重视战斗COVID-19大流行和提高设备安全协议,根据FDA的设备和放射卫生中心(CDRH)年度报告。CDRH主任杰夫博士均在9月表示,该机构是“开始拐弯”迎头赶上在上市前的审批程序和510 (k)间隙提交,专注于大流行和发行后紧急使用授权(欧洲大学协会)记录时间。
2607产品召回从去年以来最少的2013财政年度,当设备公司2304年回忆说。2013财政年度是第一个全年的FDA使这些记录。该机构公布了最多的回忆说,在3285年,在2018财年。
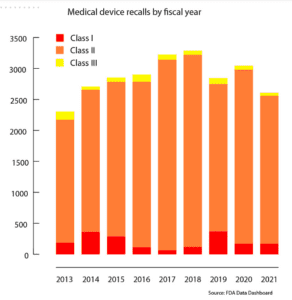 在2021财政年度,类我回忆起下降了1.2%到169,二级召回下降15%,至2389和III类下跌19.7%,至49回忆说。
在2021财政年度,类我回忆起下降了1.2%到169,二级召回下降15%,至2389和III类下跌19.7%,至49回忆说。
的最大同比变化任何类,因为FDA开始公布这些数字是在2019财年- COVID-19前的去年全年流行当类我回忆翻了两番多。我回忆起那一年,有370类(增加216.24%),2376年二级召回(下降23.5%)和97年第三类(43%)回忆说。
医疗器械召回可能是去年。但时间也看到了多年来最严重的行业之一,回忆:类我召回数百万飞利浦Respironics呼吸器,cpap BiPAPs和危险的声音消除泡沫。FDA收到超过21000医疗器械报告和124年报告的死亡人数2021年4月至4月30日,2022年。该机构要求公司提交的计划维修,更换或退款的呼吸设备错误的泡沫。检查生产设备连接到召回后,FDA说飞利浦自2015年以来已经知道泡沫问题。飞利浦已经不再接受订单的睡眠治疗系统和说它正在配合FDA以及美国司法部,传唤公司信息与召回有关。
FDA发言人吉姆·麦金尼说MDO,FDA说不确定性减少的背后是什么设备和召回事件在2021财年回忆说。
“设备召回的数量在一年内销售设备的安全的不是一个指标通常并不是一个指标,可以分析趋势,”他在一封电子邮件中说。
麦金尼说大流行“没有改变FDA的至关重要的保护和促进公共卫生监督作用,确保医疗设备是安全有效的,”但他承认COVID破坏。
“我们看到了一些中断通知和报告FDA和实现减排的努力代表的一些公司积极回忆说,“麦金尼说。“FDA将继续积极与这些企业合作,解决这些回忆说,尽快实现缓解气候变化的努力。”
 美国食品及药物管理局管理超过18000家企业生产的190000多个不同的设备在世界各地。大流行以来,FDA CDRH授予欧洲大学协会或全面营销授权超过2000医疗设备专门为COVID-19治疗,检测和诊断。
美国食品及药物管理局管理超过18000家企业生产的190000多个不同的设备在世界各地。大流行以来,FDA CDRH授予欧洲大学协会或全面营销授权超过2000医疗设备专门为COVID-19治疗,检测和诊断。
更关注流行,有更少的检查,和那些发生是虚拟的,在大多数情况下。
“这可能会导致各种各样的问题在制造、可靠性和质量方面,”迈克尔·德鲁说,医疗器械监管顾问公司和美国食品和药物管理局。
他警告从低召回数据得出结论——或庆祝他们。
“仅仅因为人们不报告并不意味着没有问题,”德鲁说。“诉讼反对医疗器械公司可能是一个更好的替代生物标志物或预后的指标如何做这个行业。”
也许我们只有一年的
从2020年3月开始,医疗保险和医疗补助服务中心(CMS)建议推迟选举程序来保护个人防护设备。CMS之后放松的指导方针,但重症监护病人的三角洲和ο变异电波进一步推迟计划过程和伤害许多医疗设备的销售。
可能会导致更少的回忆,说峡谷实验室临床研究总监大卫·洛克博士,监管科学项目教练约翰霍普金斯大学以前在强生公司监管事务的专业。
“也许我们只是做了一个好年头,产品功能好,”骆家辉说。“也许不是很多。”
设备制造商,医生和病人是FDA负责报告问题。如果一个严重问题是足够的对于一个公司启动召回,召回的机构将合作公司权衡效益相应设备的风险。这并不总是一个简单的电话。
“由美国食品和药物管理局以及公司产品责任律师和医生和病人,和其他人试图找出妥协,”德鲁说。
使困难的电话
一些设备可能保持在市场上的时间比他们应该由于害怕在流感大流行期间医疗设施短缺,说Madris金纳德,设备事件的创始人兼首席执行官、前唯一的设备标识(UDI)外部项目经理和中小企业在美国食品药品监督管理局医疗器械不良事件。
“公司正在这些决策,然后FDA可能会检查它们,但不一定足够积极的方法,“金纳德说。

BD的腋生的输液泵和生命体征监测系统旨在提供大量的流体控制,药物、血液和血液制品。照片由双相障碍。
设备的一个例子可能会造成短缺在医院如果回忆是BD的腋生的泵。2020年3月,美国食品和药物管理局呼吁全面BD召回510 (k)提交后成千上万的腋生的泵由于系统——软件和使用相关的错误。泵继续面临一系列的类我回忆,直到2021年4月。出版,BD仍有新泵的出口在美国等待所需的结果510 (k)间隙提交去年FDA。
“他们不想把他们的软件更新,如果他们被用于病人。当然,与COVID,他们可能都在使用,“金纳德说。
去年在另一个严重的设备召回,美敦力公司的HeartWare心室辅助装置(HVAD)系统设计为一座桥在心脏移植患者的风险死于晚期左心室心力衰竭,心脏组织恢复或目的地治疗在患者不能接受心脏移植。
弗里德利,一家医学技术巨头开始回忆HVAD泵植入工具在2021年3月,后来完全停止设备的销售和植入由于严重受伤或死亡的风险增加,产生市场雅培的竞争技术。
当美敦力公司停止销售HVAD 2021年6月,据报道超过100投诉涉及延误或者失败重新启动HVAD内部泵,导致14人死亡,13泵删除。医学技术巨头今年继续工作通过课堂我回忆相关设备水平。
“我认为,双相障碍腋生的泵和HVAD就是很好的例子,为什么回忆应该是强制性的,因为他们推迟,“金纳德说。“如果FDA同意因为COVID原因,或者如果有别的东西,如果它会导致市场短缺。当然,HVAD,它并没有因为有另一个公司能够更好地服务患者。”